Molecular interactions underlie nearly all biological processes, but most machine learning models treat molecules in isolation or specialize in a single type of interaction, such as protein-ligand or protein-protein binding. Here, we introduce ATOMICA, a geometric deep learning model that learns atomic-scale representations of intermolecular interfaces across five modalities, including proteins, small molecules, metal ions, lipids, and nucleic acids. ATOMICA is trained on 2,037,972 interaction complexes using self-supervised denoising and masking to generate embeddings of interaction interfaces at the levels of atoms, chemical blocks, and molecular interfaces.
ATOMICA's latent space is compositional and captures physicochemical features shared across molecular classes, enabling representations of new molecular interactions to be generated by algebraically combining embeddings of interaction interfaces. The representation quality of this space improves with increased data volume and modality diversity. As in pre-trained natural language models, this scaling law implies predictable gains in performance as structural datasets expand.
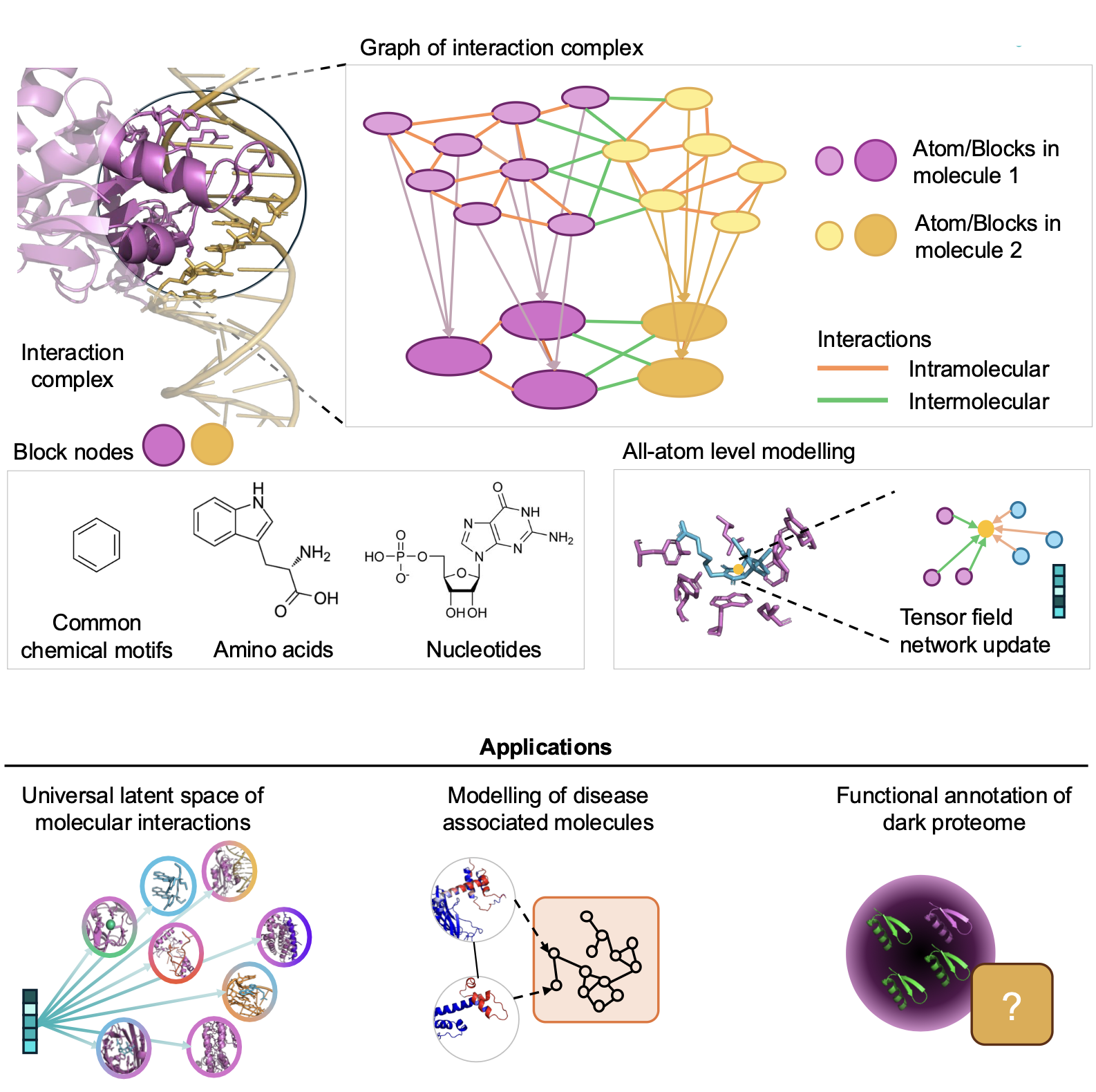
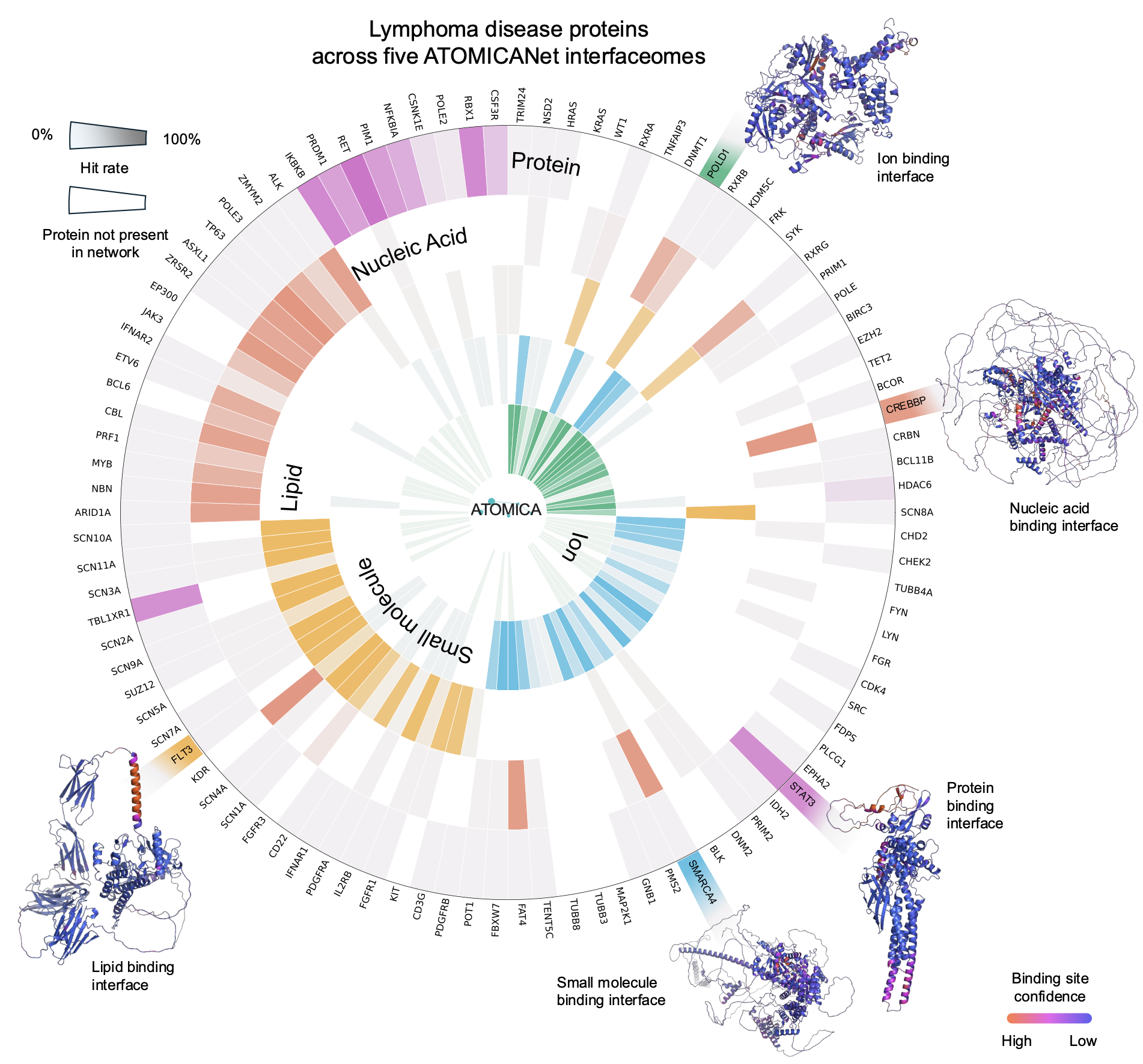
We construct modality-specific interfaceome networks, termed ATOMICANets, which connect proteins based on interaction similarity with ions, small molecules, nucleic acids, lipids, and proteins. By overlaying disease-associated proteins of 27 diseases onto ATOMICANets, we find strong associations for asthma in lipid networks and myeloid leukemia in ion networks.
We use ATOMICA to annotate the dark proteome—proteins lacking known function—by predicting 2,646 uncharacterized ligand-binding sites, including putative zinc finger motifs and transmembrane cytochrome subunits. We experimentally confirm heme binding for five ATOMICA predictions in the dark proteome. By modeling molecular interactions, ATOMICA opens new avenues for understanding and annotating molecular function at scale.
Motivation
Current machine learning models in molecular biology often treat molecules in isolation or focus narrowly on specific types of molecular interactions, such as protein-ligand or protein-protein binding. These siloed models use separate architectures for different biomolecular classes, limiting their ability to transfer knowledge across modalities. This restriction hampers generalizability and reduces performance in low-data domains like rare interactions or uncharacterized proteins.
ATOMICA addresses these limitations by unifying the modeling of intermolecular interactions across small molecules, ions, nucleic acids, peptides, and proteins. It leverages fundamental physicochemical principles common to all biomolecular interactions, such as hydrogen bonding, van der Waals forces, and π-stacking, to build a universal representation. The model is designed to operate at the atomic scale and capture multi-scale structural and chemical relationships, enabling accurate and transferable representations of molecular interfaces across diverse contexts.
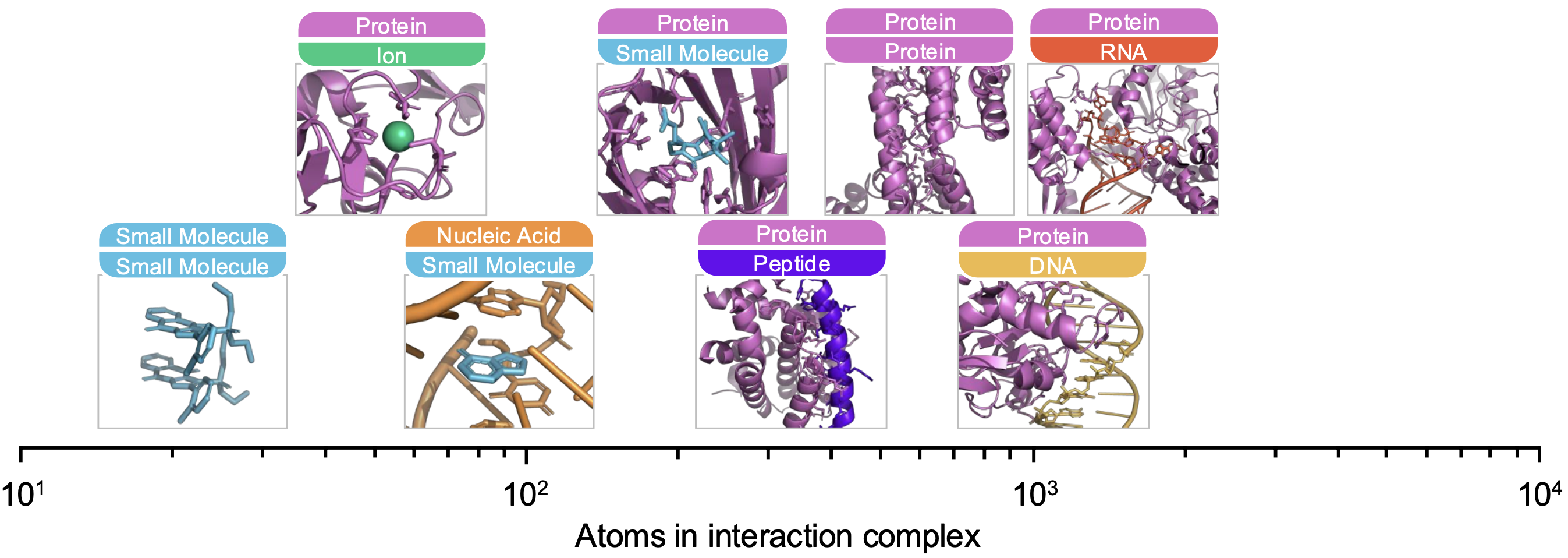
ATOMICA Model
ATOMICA is a hierarchical geometric deep learning model trained on over 2.1 million molecular interaction interfaces. It represents interaction complexes using an all-atom graph structure, where nodes correspond to atoms or grouped chemical blocks, and edges reflect both intra- and intermolecular spatial relationships. The model uses SE(3)-equivariant message passing to ensure that learned embeddings are invariant to rotations and translations of molecular structures.
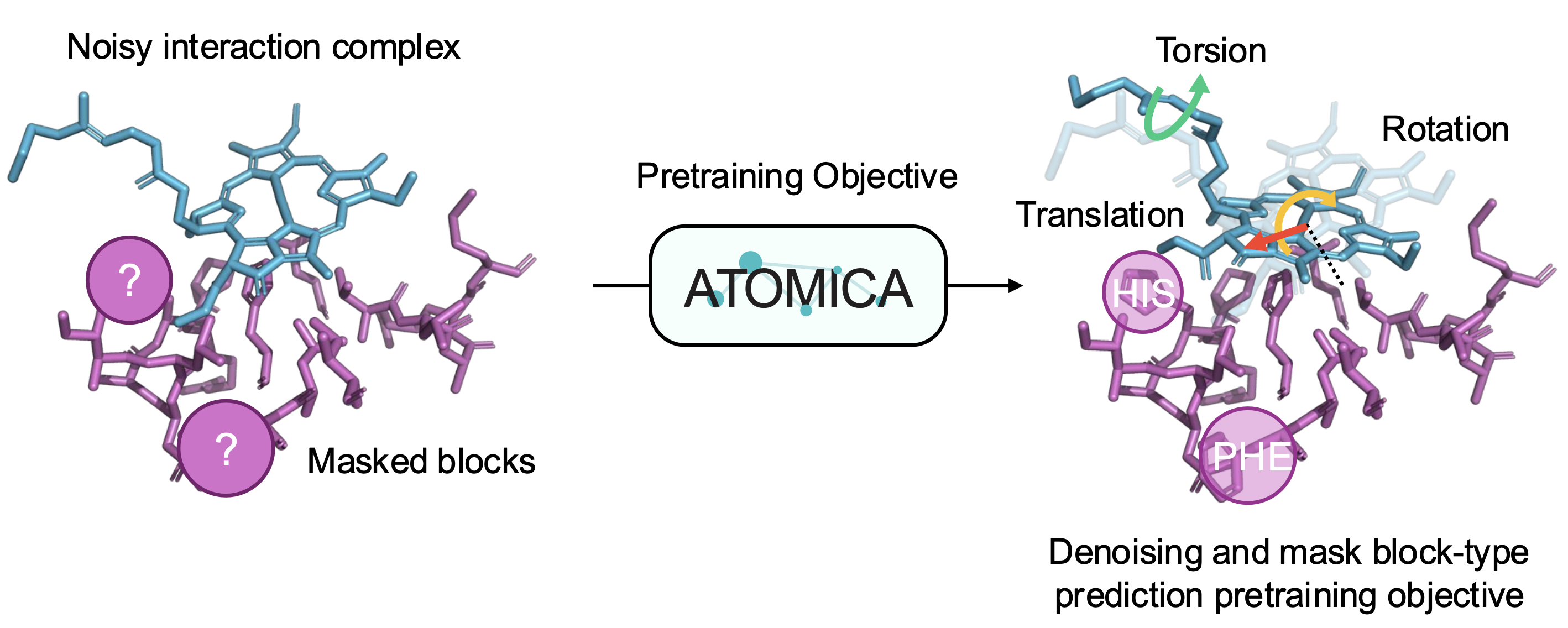
The architecture produces embeddings at multiple scales—atom, block, and graph—that capture fine-grained structural detail and broader functional motifs. The pretraining strategy involves denoising transformations (rotation, translation, torsion) and masked block-type prediction, enabling the model to learn chemically grounded, transferable features. ATOMICA supports downstream tasks via plug-and-play adaptation with task-specific heads, including binding site prediction and protein interface fingerprinting.
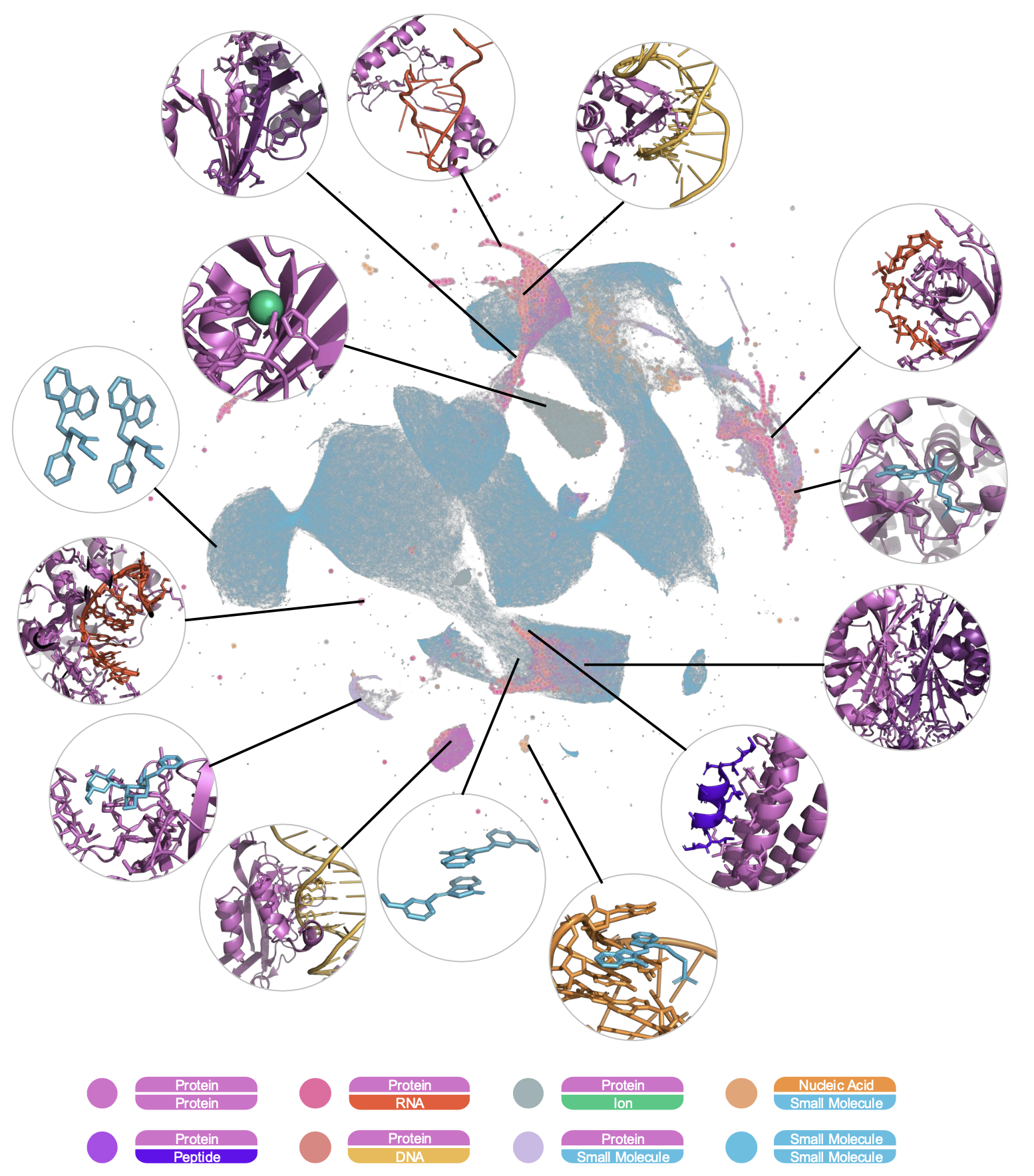
Universal Representation of Molecular Interactions
ATOMICA learns a shared latent space that encodes interaction features across all biomolecular modalities. Without explicit supervision on modality labels, the model organizes protein-protein, protein-ligand, protein-RNA, and protein-DNA interactions into chemically meaningful clusters in latent space. Embeddings show continuity across modalities—e.g., protein-peptide interactions lie close to protein-protein ones—demonstrating the model’s capacity to capture chemical and functional similarity.
This universal embedding space supports compositional reasoning, akin to word analogies in NLP. For example, the model approximates a protein-small molecule complex by algebraically combining embeddings of related complexes, reflecting learned compositionality. ATOMICA also ranks interface residues by importance using ATOMICAScore, and these predictions align well with residues involved in intermolecular bonds, outperforming sequence-only language models like ESM2 in zero-shot settings.
ATOMICA’s Interfaceome Networks
We use ATOMICA representations to construct interfaceome networks, which aregraphs linking proteins based on similarity in their interaction interfaces with ions, small molecules, nucleic acids, lipids, and proteins. These ATOMICANet modality-specific networks reveal that proteins sharing similar interface features often participate in the same disease pathways, even across different interaction types. This provides a new molecular-level view of disease mechanisms.
By analyzing disease-associated proteins across 82 diseases, ATOMICANets uncover statistically significant pathway modules in lipid, ion, and small molecule networks. The model predicts disease-relevant interactions for autoimmune neurological disorders and cancer, identifying ion channels in multiple sclerosis and lymphoma-associated proteins across all five interface types. This demonstrates ATOMICA’s value in understanding protein function and disease involvement in a modality-specific, interpretable manner.
Publication
Learning Universal Representations of Intermolecular Interactions with ATOMICA
Ada Fang, Michael Desgagné, Zaixi Zhang, Andrew Zhou, Joseph Loscalzo, Bradley L. Pentelute, and Marinka Zitnik
In Review 2025 [bioRxiv]
@article{fang2025atomica,
title={Learning Universal Representations of Intermolecular Interactions with ATOMICA},
author={Fang, Ada and Desgagné, Michael and Zhang, Zaixi and Zhou, Andrew and Loscalzo, Joseph, and Pentelute, Bradley L and Zitnik, Marinka},
journal={In Review},
url={https://www.biorxiv.org/content/10.1101/2025.04.02.646906},
year={2025}
}
Code and Data Availability
Pytorch implementation of ATOMICA is available in the GitHub repository. Datasets are also available at Harvard Dataverse repository.