With remarkable successes of machine learning in a variety of application areas, we are witnessing an increasing interest in applications of machine learning to drug discovery and development.
In this tutorial, we cover key advancements in machine learning over the last few years, with an emphasis on fundamentally new opportunities in drug development enabled by these advancements. We are interested in why and how these advances can help drug-related tasks. We elaborate uses of machine learning in drug development through six key tasks: (a) synthesis prediction and de novo drug design, (b) molecular property prediction, (c) virtual drug screening and drug-target interactions, (d) clinical trial recruitment, (e) drug repurposing, (f) adverse drug effects and polypharmacy.
We discuss theoretical foundations behind methods for these key drug-related tasks, illustrate various approaches based on different formulations, and summarize representative applications. We cover generative models, reinforcement learning, as well as very recent advancements in deep representation learning and embeddings. In doing so, we present a toolbox of AI algorithms for end-to-end drug development.
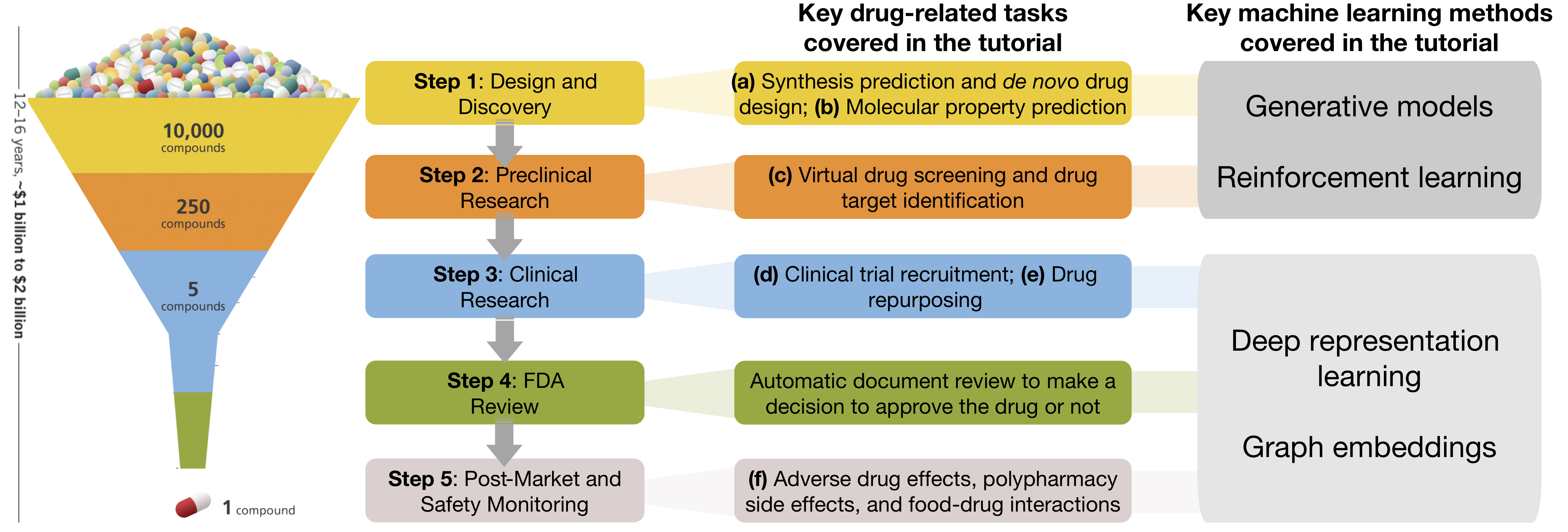
Overview
Drug discovery and development is a long and expensive process. It usually starts with experimental discovery of molecules and targets (i.e., de novo drug design), and validation of discoveries with in vitro experiments on cell lines, organoids, and animals before moving to clinical testing. The entire process from discovery to the regulatory approval of a new drug can take as much as 12 years and cost upwards of US$2.8 billion. Furthermore, huge uncertainty (1:5000 success rate) is associated with each drug development stage.
Machine learning methods have emerged as a promising tool to address these challenges and accelerate drug development. In the tutorial, we cover the following key drug-related tasks:
- Synthesis prediction and de novo drug design (i.e., designing an entirely new molecule from scratch) aims to generate chemically correct structures to assist in complex molecule synthesis.
- Molecular property prediction aims to identify therapeutic effects of molecules by predicting properties, such as potency, bioactivity, and toxicity, from the molecular data.
- Virtual drug screening and drug target identification aim to predict how drugs affect the human body by binding to target proteins and affecting their downstream activity.
- Clinical trial recruitment aims to identify the right doctors to help conduct the trials as well as find qualified patients to participate the trials.
- Drug repurposing seeks to find new uses for known drugs as well as for novel molecules through the use of drug chemical, target and side effect similarity between drugs and diseases.
- Adverse drug effects, polypharmacy, and drug-food interaction prediction aims to predict mechanisms causing adverse drug effects, suggest alternative drug members for the intended pharmacological effects without negative health effects, and predict the effects of food constituents on interacting drugs.
We then discuss key classes of methods for tackling these drug-related tasks:
- Generative models. We focus on variational autoencoders (VAE) and generative adversarial networks (GAN) that are well suited for de novo molecule design. They take as input line or graph-based compound representations with known therapeutic properties, encode the compounds into latent spaces, and then decode them into new drug samples.
- Reinforcement learning. We mainly talk about policy gradient methods, state-of-the-art methods for molecule generation that can incorporate domain-specific knowledge about molecule synthesis.
- Deep representation learning. We present major neural architectures for learning representations of drug-related data. These methods are relevant because they achieve state-of-the-art performance on drug-related tasks. For example, the methods were used to automatically learn drug fingerprints, learn drug-protein binding affinity, and recruit patients into clinical trials. Further, graph embedding methods are used to study drug combinations and predict drug effects as they spread throughout biological networks beyond the molecules to which they directly bind.
Program
- (15 min): Overview and introduction [Slides]
- (30 min): Virtual drug screening, knowledge graph embedding methods for drug repurposing in rare and emerging diseases [Slides]
- (30 min): Adverse drug effects, drug-drug interactions, active learning for modeling of drug combinations [Slides]
- (30 min): Clinical trial site identification (doctor identification, covid vaccine and antibody trial site identification), patient recruitment [Video]
- (30 min): Molecule optimization, molecular graph generation, multimodal graph-to-graph translation [Video Part 1] [Video Part 2] [Video Part 3]
- (30 min): Molecular property prediction, transformers for prediction of molecular interactions [Video Part 1] [Video Part 2] [Video Part 3] [Video Part 4]
- (15 min): Hands-on exercise with demos, implementation details, tools, and tips [Slides]
- (15 min): Wrap-up and Q&A session [Slides]
Tutorial materials
The tutorial slides will be posted on this website and made available to all participants. Demos and hands-on exercises will use datasets and code available in Therapeutics Data Commons (TDC). We encourage participants to explore tutorials that are included in the TDC Github repository and DeepPurpose Github repository. We also provide an interactive user interface MolDesigner that provides support for design of efficacious drugs with deep learning.
Tutorial info
The tutorial will be held at the IJCAI conference, January 7-15, 2021 online in a virtual reality. The tutorial will take place on January 7, 2021, 9am-12:15pm JST (Japanese Standard Time) / January 6, 2021 7:00-10:15pm EST (Eastern Standard Time).
The target audience for this tutorial are participants with knowledge of the fundamentals of machine learning and some experience in deep learning (Intermediate). No special software or other package installation is needed to follow this tutorial.
Presenters
Marinka Zitnik is Assistant Professor at Harvard, Associate Member at the Broad Institute of MIT and Harvard and Faculty at Harvard Data Science. Dr. Zitnik is a computer scientist studying applied machine learning with a focus on challenges brought forward by data in science, medicine, and health. Before Harvard, she was a postdoc in Computer Science at Stanford. She received her Ph.D. in Computer Science from University of Ljubljana while also researching at Imperial College London, University of Toronto, Baylor College of Medicine, and Stanford. Her work received several best paper, poster, and research awards from the International Society for Computational Biology. Her research recently won the Bayer Early Excellence in Science Award. She was named a Rising Star in Electrical Engineering and Computer Science (EECS) by MIT and also a Next Generation in Biomedicine by Broad Institute of MIT and Harvard, being the only young scientist who received such recognition in both EECS and Biomedicine. She has published in top ML venues (e.g., NeurIPS, ICLR) and top journals (e.g., Nature Communications, Proceedings of the National Academy of Sciences, PNAS), co-organized a tutorial in the area at ISMB 2018, a related workshop at ICLR 2019, gave invited talks on this topic in big pharma and at major conferences.
Cao (Danica) Xiao is the Director of Machine Learning at Analytics Center of Excellence of IQVIA. She is leading IQVIA’s North America machine learning team to drive next generation healthcare AI. Her team works on various projects on disease prediction, in silico drug modeling (e.g., adverse drug reaction detection, drug repositioning and de novo design) and clinical trial recruitment prediction. Her research focuses on using ML/AI approaches to solve diverse real world healthcare challenges. Particularly, she is interested in phenotyping on electronic health records, data mining for in-silico drug modeling, patient segmentation for neuro-degenerative diseases. Her research has been published in leading AI conferences including KDD, NIPS, ICLR, AAAI, IJCAI, SDM, ICDM, WWW and top health informatics journals such as Nature Scientific Reports and JAMIA. Prior to IQVIA, she was a research staff member in the AI for Healthcare team at IBM Research from 2017 to 2019 and served as member of the IBM Global Technology Outlook Committee from 2018 to 2019. She acquired her Ph.D. degree from University of Washington, Seattle in 2016.
Jimeng Sun is Professor in the Computer Science Department at University of Illinois Urbana-Champaign (UIUC). Prior to UIUC, he worked at Georgia Tech and IBM Research. His research is on artificial intelligence (AI) for healthcare. And the core topics include 1) Deep learning for drug discovery, 2) Clinical trial optimization, 3) Computational phenotyping, 4) Clinical predictive modeling, 5) Treatment recommendation and 6) Health monitoring. Dr. Sun has been collaborating with many healthcare organizations. He published over 200 papers with h-index 59 and filed over 20 patents. He has received SDM/IBM early career research award 2017, ICDM best research paper award in 2008, SDM best research paper award in 2007, and KDD Dissertation runner-up award in 2008. In 2019, he was recognized as Top 100 AI Leaders in Drug Discovery and Advanced Healthcare. Dr. Sun received B.S. and M.Phil. in Computer Science from Hong Kong University of Science and Technology in 2002 and 2003, M.Sc and PhD in Computer Science from Carnegie Mellon University in 2006 and 2007.
Postdoctoral research fellows in knowledge graphs and graph ML
Prof. Marinka Zitnik invites applications for Postdoctoral Research Fellowships at Harvard University.
We have an opening for a postdoctoral research fellowship in novel methods for knowledge graphs and graph representation learning. This position in available immediately. We are currently reviewing applications for this position. Interested candidates are encouraged to submit their applications as soon as possible.
Therapeutics Data Commons
