Abstract
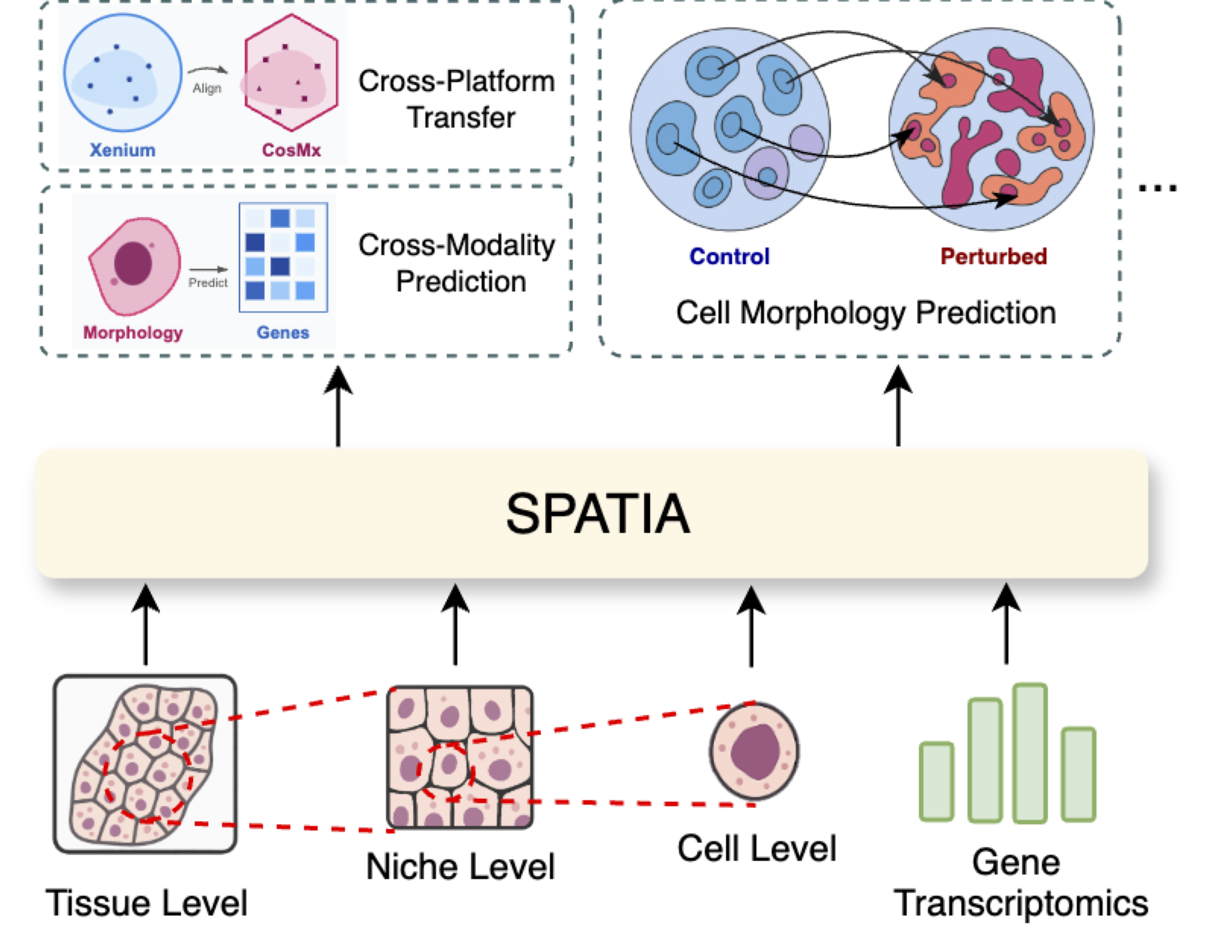
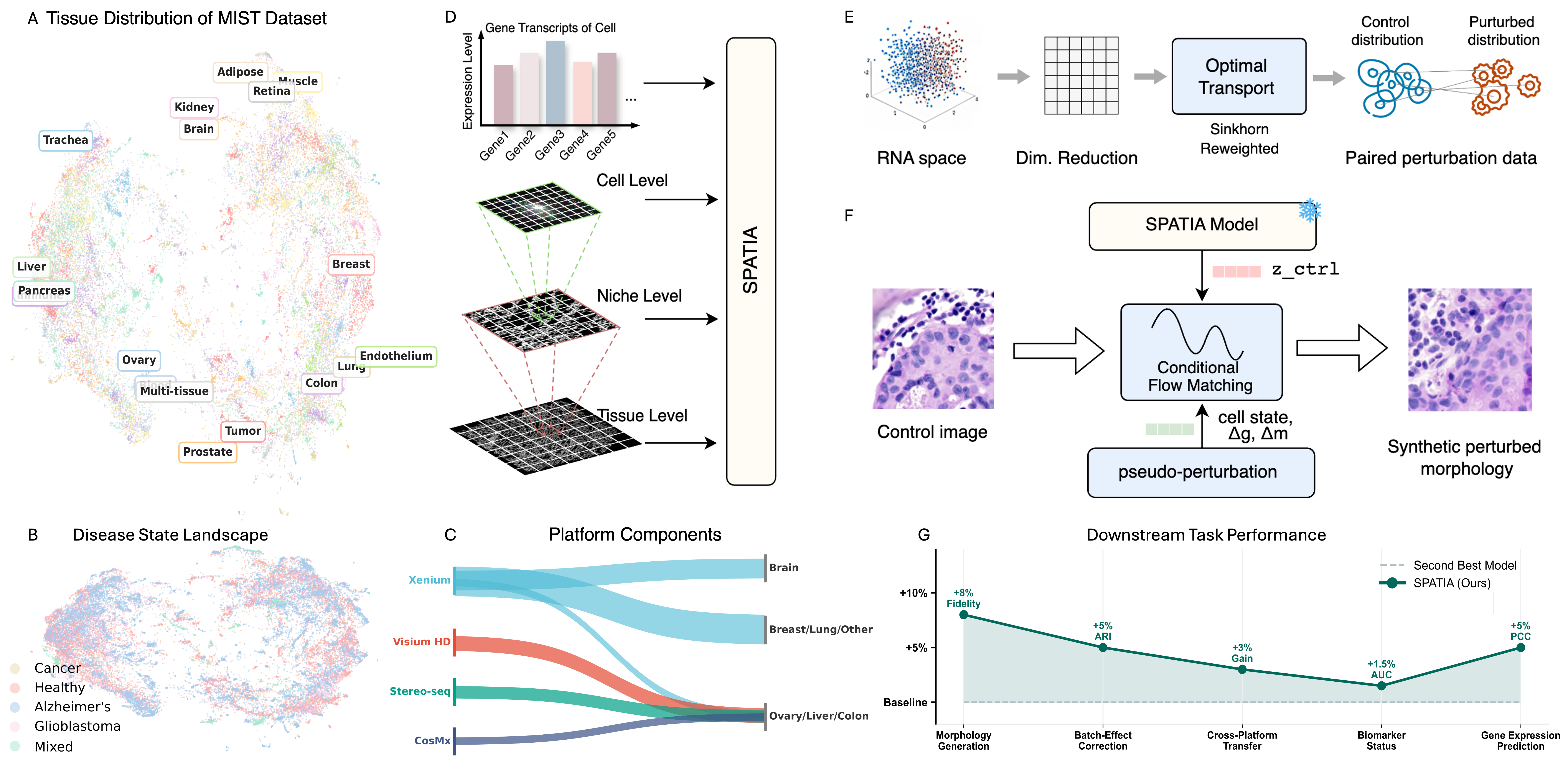
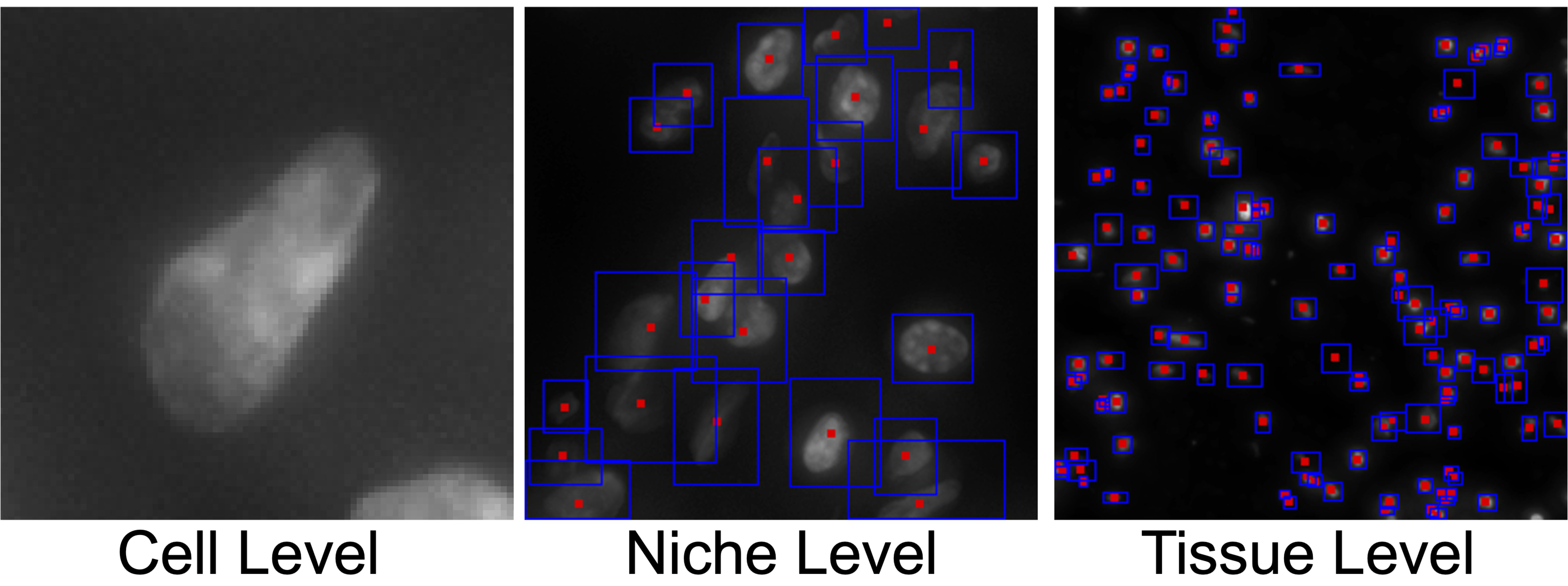
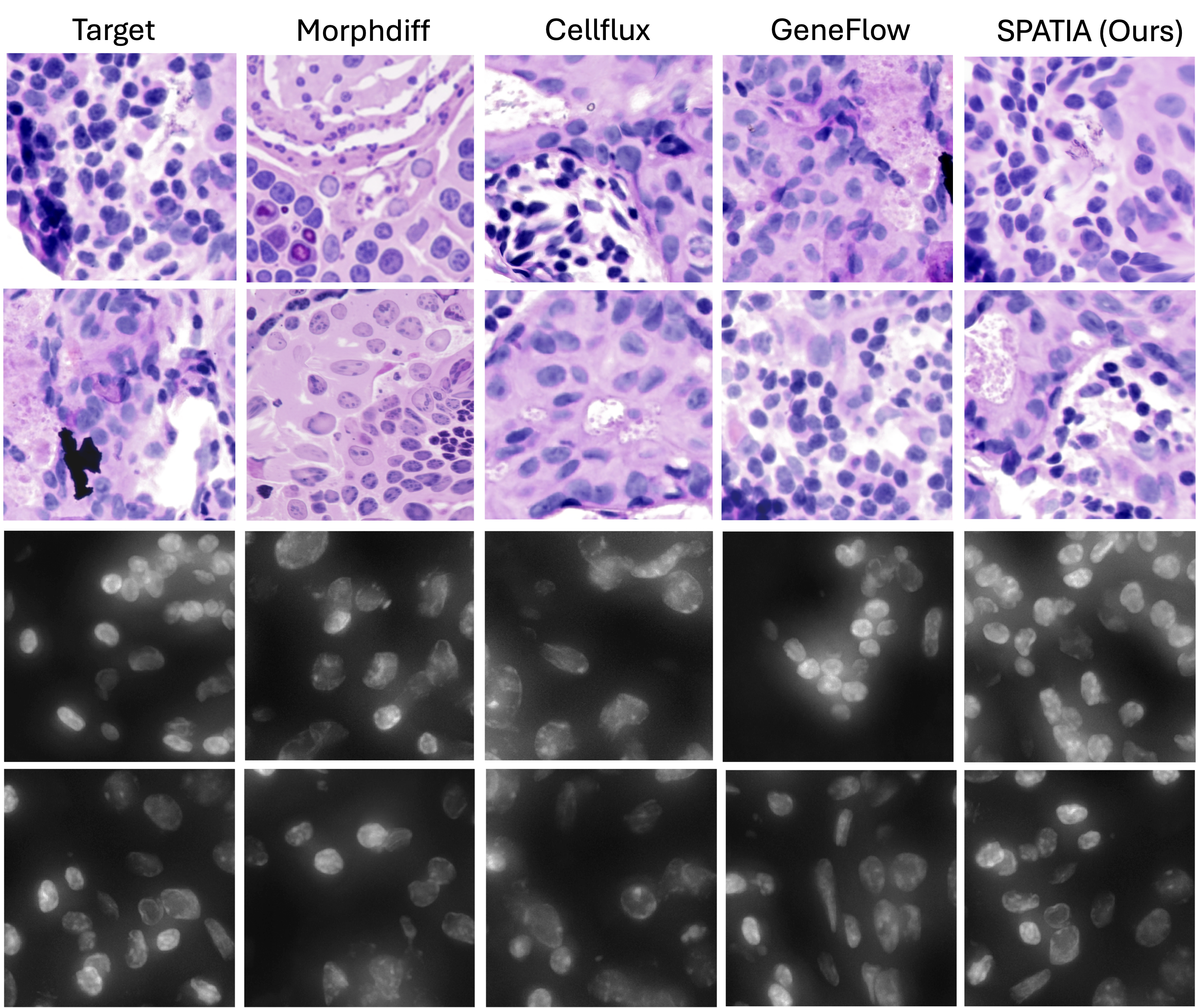
Understanding how cellular morphology, gene expression, and spatial context jointly shape tissue function is a central challenge in biology. Image-based spatial transcriptomics technologies now provide high-resolution measurements of cell images and gene expression profiles, but existing methods typically analyze these modalities in isolation or at limited resolution. We address the problem by introducing SPATIA, a multi-level generative and predictive model that learns unified, spatially aware representations by fusing morphology, gene expression, and spatial context from the cell to the tissue level. SPATIA also incorporates a novel spatially conditioned generative framework for modeling target-state morphology distributions under observed biological transition families. Specifically, we propose a confidence-aware flow matching objective that reweights weak optimal-transport pairs based on uncertainty. We further apply morphology-profile alignment to encourage biologically meaningful image generation, enabling the modeling of microenvironment-dependent phenotypic transitions. We assembled a multi-scale dataset consisting of 25.9 million cell-gene pairs across 17 tissues. We benchmark SPATIA against 18 models across 12 tasks, spanning categories such as phenotype generation, annotation, clustering, gene imputation, and cross-modal prediction. SPATIA achieves improved performance over state-of-the-art models, improving generative fidelity by 8% and predictive accuracy by up to 3%.